
LRRK2 activation in idiopathic Parkinson’s disease
New proximity ligation assays were developed and validated
As a LRRK2 autophosphorylation site, pSer1292 reflects the activity of LRRK2 per se. We developed a proximity ligation assay to amplify the signal and increase the specificity of an antibody recognizing the pSer1292 epitope of the LRRK2 protein. For the proximity ligation assay, we paired the anti-pSer1292 antibody with a validated antibody directed against an epitope in the C terminus of Roc (COR) domain of the LRRK2 protein. In this way, the signal of the anti-pSer1292 antibody was amplified and detected only if it was in proximity to the anti-COR domain antibody (that is, specific LRRK2 pSer1292 signals were amplified, whereas potential signals from nonspecific or off-target antibody binding were filtered out by the proximity ligation assay). We developed a second proximity ligation assay that assessed the interaction between LRRK2 and 14-3-3 proteins, whose binding to LRRK2 is associated with reduced kinase activity. The proximity ligation assays were designed such that LRRK2 activity would be associated with a strong pSer1292 signal and a weak 14-3-3 signal; conversely, when LRRK2 is inactive, there would be little pSer1292 signal and a robust 14-3-3 signal.To validate the assays, we used wild-type LRRK2, mutant LRRK2 (LRRK2G2019S/G2019S), and LRRK2-deficient (LRRK2−/−) human embryonic kidney–293 (HEK-293) cells, obtained by CRISPR/Cas9 gene editing. In wild-type cells, there was little pSer1292 proximity ligation assay signal and a strong 14-3-3 signal (Fig. 1, A, B, D, and E). The G2019S mutation is known to cause increased LRRK2 kinase activity (5, 6). Accordingly, in LRRK2G2019S/G2019S HEK-293 cells, there was a bright pSer1292 signal and loss of the 14-3-3 interaction [P < 0.0001 for both compared to wild-type cells; analysis of variance (ANOVA) with Bonferroni correction]. No signal in either proximity ligation assay was seen in LRRK2−/− HEK-293 cells lacking LRRK2 (Fig. 1, A, B, D, and E), further establishing the specificity of the assays. The small GTPase, Rab10, has recently been shown to be directly phosphorylated at Thr73 by LRRK2 (6). Using a specific pThr73-Rab10 antibody, we found low amounts of phosphorylated Rab10 in wild-type cells and much higher amounts in LRRK2G2019S/G2019S cells (P < 0.0001 compared to wild-type cells), in keeping with increased kinase activity of the mutant protein (Fig. 1, A, C, and D). After assay development, validation included blinded analysis and correct identification of all three cell lines with these assays alone. Using the selective LRRK2 kinase inhibitors, GNE-7915 and MLi-2 (13), in dose-response studies, we found that LRRK2 activation state, assessed by the pSer1292 signal in the proximity ligation assay, correlated closely with phosphorylation of its substrate Rab10 (Fig. 1, F and G). We next looked at LRRK2 activation state in patient-derived lymphoblastoid cell lines. Relative to cells derived from a healthy age-matched control, there was marked elevation of the pSer1292 signal in a LRRK2 G2019S mutation carrier; titration with the selective LRRK2 kinase inhibitor, GNE-7915, dose-dependently reduced the pSer1292 proximity ligation signal and pThr73-Rab10 signal in parallel (Fig. 1H).
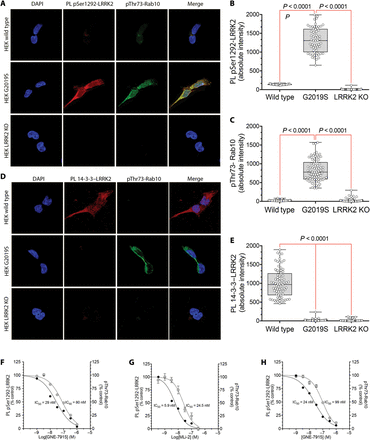
Fig. 1 Validation of proximity ligation assays using CRISPR/Cas9 gene-edited HEK-293 cells and LRRK2 kinase inhibitors.(A) A proximity ligation (PL) assay showing LRRK2 kinase activation by means of phosphorylation of the autophosphorylation site Ser1292 (red signal) and immunofluorescence of phosphorylation of the LRRK2 substrate Rab10 (green signal). In wild-type HEK-293 cells (HEK wild type; top row), there was little proximity ligation signal or pThr73-Rab10 immunofluorescence. HEK-293 cells carrying a homozygous G2019S mutation in LRRK2 (HEK G2019S; middle row) showed elevated LRRK2 kinase activity, indicated by a bright pSer1292 proximity ligation signal and strong pThr73-Rab10 immunofluorescence. In HEK-293 cells lacking LRRK2 [HEK LRRK2 knockout (KO; bottom row)], there was no pSer1292 proximity ligation signal and very little pThr73-Rab10 signal. DAPI (4′,6-diamidino-2-phenylindole; blue) was used as a nuclear stain. (B) Quantification of the pSer1292 proximity ligation signal in wild-type, G2019S mutant, and knockout HEK-293 cells. Results reflect three independent experiments. Each symbol represents signal from a single cell. Statistical testing by ANOVA with post hoc Bonferroni correction. (C) Quantification of the pThr73-Rab10 signal in wild type, G2019S mutant, and knockout HEK-293 cells. Results reflect three independent experiments. Each symbol represents signal from a single cell. Statistical testing by ANOVA with post hoc Bonferroni correction. (D) Proximity ligation assay of 14-3-3 binding to LRRK2 and immunofluorescence of Rab10 phosphorylation at Thr73. In wild-type HEK-293 cells (top row), there was a strong 14-3-3–LRRK2 proximity ligation signal (red) and little pThr73-Rab10 immunofluorescence (green). In HEK-293 cells carrying a homozygous G2019S mutation in LRRK2 (middle row), there was loss of 14-3-3 binding and a marked increase in pThr73-Rab10 signal. In HEK-293 LRRK2 knockout cells (bottom row), there was no 14-3-3–LRRK2 signal and little pThr73-Rab10 signal. (E) Quantification of the 14-3-3–LRRK2 proximity ligation signal in HEK-293 wild type, G2019S mutant, and LRRK2 knockout cells. Results reflect three independent experiments. Each symbol represents signal from a single cell. Statistical testing by ANOVA with post hoc Bonferroni correction. (F) Dose-response curves for the LRRK2 kinase inhibitor GNE-7915 against the pSer1292 proximity ligation signal (filled circles) and the pThr73-Rab10 signal (open circles) in HEK-293 G2019S mutant cells. Cells were cultured for 24 hours with various LRRK2 kinase inhibitor concentrations. Results are from three independent experiments. Symbols show means ± SEM. IC50 values were calculated by GraphPad Prism software. (G) Dose-response curves for the LRRK2 kinase inhibitor MLi-2 against the pSer1292 proximity ligation signal (filled circles) and the pThr73-Rab10 signal (open circles) in HEK-293 G2019S mutant cells. Cells were cultured for 24 hours with various LRRK2 kinase inhibitor concentrations. (H) Dose-response curves for the LRRK2 kinase inhibitor GNE-7915 against the pSer1292 proximity ligation signal (filled circles) and the pThr73-Rab10 signal (open circles) in lymphoblastoid cells derived from an individual carrying the G2019S LRRK2 mutation. Cells were cultured for 24 hours with various LRRK2 kinase inhibitor concentrations.
Endogenous wild-type LRRK2 is activated in iPD
Conventional assays of LRRK2 activity often rely on overexpression, and they require substantial amounts of tissue, lack cellular/anatomical resolution, and cannot be performed in previously fixed tissue. In contrast, our proximity ligation assays could assess the activation state of endogenous LRRK2 on a cell-by-cell basis using fixed cells or tissue. In this context, we measured pSer1292 proximity ligation and pThr73-Rab10 by quantitative confocal immunofluorescence in sections of substantia nigra from postmortem brain tissue from seven individuals who had died with iPD and from eight controls matched for age and postmortem interval. In nigrostriatal dopamine neurons from healthy controls, there were very low basal levels of pSer1292 signal and pThr73-Rab10 immunoreactivity and a strong 14-3-3 proximity ligation signal (Fig. 2). In contrast, the remaining nigrostriatal dopamine neurons of the iPD cases showed about a sixfold increase in pSer1292 proximity ligation (P < 0.0002, two-tailed, unpaired t test; P < 0.002 with Welch’s correction for unequal variances), and this was associated with a fourfold increase in phosphorylation (pThr73) of the LRRK2 substrate Rab10 (P < 0.0002). The increase in LRRK2 activation state in iPD dopamine neurons corresponded to a fivefold decrease in the 14-3-3 proximity ligation signal (P < 0.0001; P < 0.0004 with Welch’s correction). This suggested that endogenous wild-type LRRK2 may be activated in dopamine neurons in iPD and that this activation was associated with increased substrate phosphorylation.
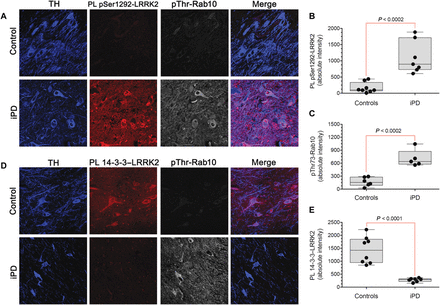
Fig. 2 Activation of LRRK2 kinase in nigrostriatal dopamine neurons in human iPD postmortem brain tissue.(A) Shown are the pSer1292 proximity ligation signal (red) and pThr73-Rab10 immunofluorescence signal (gray) in sections of substantia nigra from a healthy, age-matched control human brain (top row) and a brain from an individual with iPD (bottom row). In the control brain, there was little pSer1292 or pThr73-Rab10 signal, but in the iPD brain, there were strong signals for both. TH, tyrosine hydroxylase, a marker of dopamine neurons (blue). (B) Quantification of pSer1292 proximity ligation signal in eight control brains and seven iPD brains. Statistical comparison by unpaired two-tailed t test. (C) Quantification of pThr73-Rab10 signal in eight control brains and seven iPD brains. Statistical comparison by unpaired two-tailed t test. (D) Shown are 14-3-3–LRRK2 proximity ligation signal (red) and pThr73-Rab10 immunofluorescence signal (gray) in sections of substantia nigra from a control human brain (top row) and a brain from an individual with iPD (bottom row). In the control brain, there was a strong 14-3-3–LRRK2 proximity ligation signal and little pThr73-Rab10 signal, but in the iPD brain, the opposite pattern was seen. (E) Quantification of 14-3-3–LRRK2 proximity ligation signal in eight control brains and seven iPD brains. Statistical comparison by unpaired two-tailed t test.In addition to neuronal expression, LRRK2 is expressed by microglia (14). We found detectable levels of pSer1292 proximity ligation signal in nigral microglia in controls (fig. S1, A and B), and the signal was more than doubled in microglia from iPD cases (P < 0.0005, two-tailed, unpaired t test).
Endogenous wild-type LRRK2 activation is found in the rotenone (mitochondrial) and α-synuclein rat models of PD
Mitochondrial impairment and α-synuclein aggregation and accumulation have been strongly implicated in PD pathogenesis (15). Therefore, we tested whether LRRK2 was activated in two rat models of PD. First, we used the rotenone model of PD, which has been shown to reproduce or even predict pathological and pathogenic features of the disease (16). Using substantia nigra sections from the brains of rotenone-treated rats that had reached behavioral endpoint (severe parkinsonism after 10 to 14 days), we found a 10-fold increase in pSer1292 proximity ligation signal in nigrostriatal dopamine neurons compared to vehicle-treated control rat nigrostriatal dopamine neurons (P < 0.0001, unpaired, two-tailed t test) (Fig. 3, A to C). In these animals, there was a marked loss of the 14-3-3 proximity ligation signal (P < 0.0001, unpaired, two-tailed t test) similar to the changes we observed in human postmortem brain tissue from iPD patients. We found detectable pSer1292 proximity ligation signal in microglia in vehicle-treated control rat brain (fig. S1, C and D); the signal was more than doubled after rotenone treatment of rats (P < 0.05, unpaired, two-tailed t test). To determine whether LRRK2 activation occurs before neurodegeneration, we examined tissue from animals that had received only 1 or 5 days of rotenone treatment, time points at which we detect no degeneration of the nigrostriatal dopamine neurons (fig. S2). After a single dose of rotenone, the pSer1292 proximity ligation signal was increased fivefold relative to vehicle treatment (P < 0.0001, ANOVA with Bonferroni correction), and after five daily doses, the proximity ligation signal was increased sixfold (P < 0.0001).Fig. 3 LRRK2 activation in nigrostriatal dopamine neurons in two rat models of PD.(A) Shown are pSer1292 and 14-3-3–LRRK2 proximity ligation signals in the substantia nigra of the brains of rats treated with vehicle (control, top row) or the pesticide rotenone (bottom row). In the rotenone-treated rats, there was increased pSer1292 proximity ligation signal and loss of 14-3-3–LRRK2 proximity ligation signal, indicating LRRK2 activation. TH, tyrosine hydroxylase, a marker of dopamine neurons (blue). (B) Quantification of pSer1292 proximity ligation signal in nigrostriatal dopamine neurons from control vehicle- and rotenone-treated rats. Symbols represent individual animals. Statistical comparison by unpaired two-tailed t test. (C) Quantification of 14-3-3–LRRK2 proximity ligation signal in nigrostriatal dopamine neurons from control vehicle- and rotenone-treated rats. Symbols represent individual animals. Statistical comparison by unpaired two-tailed t test. (D) Shown are pSer1292 proximity ligation signal and 14-3-3–LRRK2 proximity ligation signal in the substantia nigra of the brains of rats that received a unilateral injection of AAV2-hSNCA into one brain hemisphere. In the hemisphere overexpressing α-synuclein (bottom row), there was increased pSer1292 proximity ligation signal and loss of 14-3-3–LRRK2 proximity ligation signal, indicating LRRK2 activation in nigrostriatal neurons compared to the hemisphere that was not injected (top row). (E) Quantification of pSer1292 proximity ligation signal in nigrostriatal dopamine neurons from the control and AAV-hSNCA–injected rat brain hemispheres. Symbols represent mean values from each hemisphere. Statistical comparison by paired two-tailed t test. (F) Quantification of 14-3-3–LRRK2 proximity ligation signal in nigrostriatal dopamine neurons from the control and AAV-hSNCA–injected rat brain hemispheres. Symbols represent mean values from each hemisphere. Statistical comparison by paired two-tailed t test.α-Synuclein accumulation and Lewy body pathology are hallmarks of PD. Elevated wild-type α-synuclein may cause PD, and several groups have used viral vector–mediated overexpression of α-synuclein as a model of PD (17). Here, we used adeno-associated virus type 2 (AAV2)–mediated overexpression of wild-type human α-synuclein (hSNCA) injected unilaterally into the substantia nigra pars compacta of rats to induce slowly progressive neurodegeneration. Six weeks after vector injection when neurodegeneration was ongoing, the remaining nigrostriatal dopamine neurons showed a marked 10-fold increase in pSer1292 proximity ligation signal compared to the contralateral, uninjected hemisphere (P < 0.0001, paired two-tailed t test) (Fig. 3, D to F); there was a concomitant loss of the 14-3-3 proximity ligation signal (P < 0.0001).Both rotenone treatment and AAV2-mediated α-synuclein overexpression lead to oligomerization of α-synuclein, as well as accumulation of Ser129-phosphorylated α-synuclein (18, 19). We recently identified soluble oligomers and pSer129-α-synuclein as specific forms of α-synuclein that have deleterious effects on mitochondrial protein import machinery and that cause mitochondrial impairment (15). In analogous fashion, when SNCA−/− HEK-293 cells were treated with exogenous soluble oligomers of α-synuclein (400 nM monomer equivalent) for 24 hours, there was a marked activation of endogenous wild-type LRRK2 with increased pSer1292 and loss of the 14-3-3 proximity ligation signal (P < 0.0001, ANOVA with Bonferroni correction; fig. S3). Treatment with monomeric α-synuclein did not activate LRRK2.Both rotenone treatment and elevated α-synuclein increase formation of reactive oxygen species (ROS), and both insults activate wild-type LRRK2, which raises the possibility that it is secondary generation of ROS that actually activates LRRK2. To test directly whether ROS can activate LRRK2, we treated wild-type HEK-293 cells with H2O2 (Fig. 4, A and B). Treatment with H2O2 dose-dependently (50 nM to 5 μM) activated the pSer1292 proximity ligation signal (P < 0.0001 versus control for all H2O2 doses; one-way ANOVA with Bonferroni correction) and increased phosphorylation of its substrate Rab10 (P < 0.0001 for all doses above 50 nM H2O2). The antioxidant α-tocopherol blocked H2O2 activation of the pSer1292 signal (P < 0.0001) and Rab10 phosphorylation (P < 0.0001).
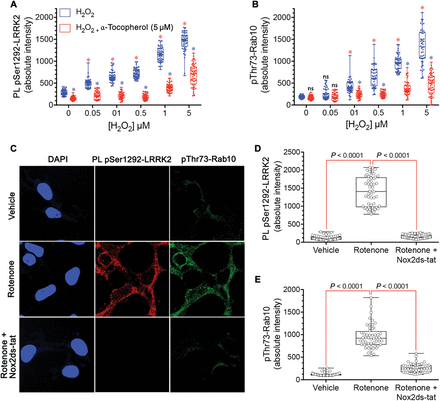
Fig. 4 LRRK2 is activated in HEK-293 cells by ROS.(A) The pSer1292 proximity ligation signal is increased dose-dependently by H2O2 (blue symbols) in wild-type HEK-293 cells. This H2O2-induced increase was blocked by the antioxidant α-tocopherol (5 μM) (red symbols). Results represent three independent experiments. Symbols represent measurements from individual cells. Red asterisks denote P < 0.0001 versus no H2O2, ANOVA with Bonferroni correction; blue asterisks denote P < 0.0001 versus H2O2 alone at the same concentration. (B) pThr73-Rab10 signal was increased dose-dependently by H2O2 (blue symbols) in wild-type HEK-293 cells, and the H2O2-induced increase was blocked by the antioxidant α-tocopherol (5 μM) (red symbols). Results represent three independent experiments. Symbols represent measurements from individual cells. Red asterisks denote P < 0.0001 versus no H2O2, ANOVA with Bonferroni correction; blue asterisks denote P < 0.0001 versus H2O2 alone at the same concentration. ns, not significant. (C) In wild-type HEK-293 cells, rotenone treatment increased the pSer1292 proximity ligation signal and pThr73-Rab10 immunoreactivity. Both effects were blocked by the specific NOX2 inhibitor Nox2ds-tat. (D) Quantification of the pSer1292 proximity ligation signal in vehicle- and rotenone-treated cells. Results represent three independent experiments. Symbols represent measurements from individual cells. Comparison by ANOVA with Bonferroni correction. (E) Quantification of the pThr73-Rab10 immunofluorescence signal in vehicle- and rotenone-treated cells. Results represent three independent experiments. Symbols represent measurements from individual cells. Comparison by ANOVA with Bonferroni correction.Further evidence of oxidative activation of LRRK2 came from the study of endogenous NADPH oxidase 2 (NOX2). We found that rotenone treatment of wild-type HEK-293 cells caused an increase in the pSer1292 proximity ligation signal and Rab10 phosphorylation (Fig. 4, C to E). Although rotenone may cause mitochondrial ROS formation, mitochondrially derived ROS may also activate NOX2 in a process known as ROS-induced ROS release, which can feed forward to amplify ROS production (20, 21). We found that cotreatment with rotenone plus the specific NOX2 inhibitor peptide, Nox2ds-tat (22), blocked rotenone’s effects on LRRK2 activation and phosphorylation of its substrate (P < 0.0001, one-way ANOVA with Bonferroni correction). Thus, NOX2-generated superoxide appears to be important in activating LRRK2.
A LRRK2 kinase inhibitor prevents rotenone-induced activation of nigrostriatal LRRK2 and its downstream effects in rats
The rotenone model of PD reproduces many features of the human disease, including accumulation of pSer129-α-synuclein, impairment of autophagy, and reduced mitochondrial protein import (15). To determine whether systemic treatment with a brain-penetrant LRRK2 inhibitor could block rotenone-induced LRRK2 activation and to survey some of the potential downstream effects of LRRK2 activation, we treated rats for 5 days with rotenone (2.8 mg/kg per day, i.p.) with or without concomitant PF-360 (10 mg/kg, p.o., twice daily), a highly selective LRRK2 kinase inhibitor (23, 24). This PF-360 dosing regimen resulted in a pharmacokinetic profile in which an IC90 concentration in rat brain was achieved for 15 hours daily, and an IC50 concentration was achieved for a full 24 hours.In a new cohort of rats treated with rotenone for 5 days, there was a marked increase in pSer1292 proximity ligation signal in nigrostriatal dopamine neurons, which was associated with an increase in phosphorylation of Rab10 (Fig. 5, A to C). Cotreatment with PF-360 effectively blocked the rotenone-induced activation of LRRK2 (P < 0.0001, two-way ANOVA with Sidak correction) and phosphorylation of Rab10 (P < 0.0001). Thus, the pSer1292 proximity ligation assay provided an ex vivo assay of target (LRRK2) engagement by PF-360, which was corroborated by measurement of pThr73-Rab10.
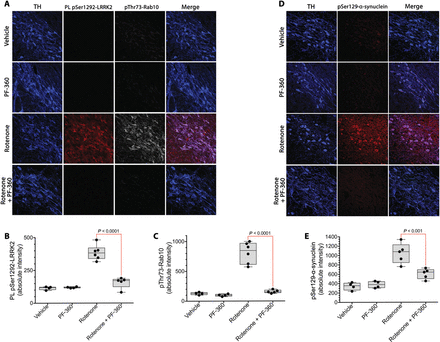
Fig. 5 LRRK2 activation and pSer129-α-synuclein accumulation in rat nigrostriatal dopamine neurons can be blocked by a brain penetrant LRRK2 kinase inhibitor.(A) Shown are the pSer1292 proximity ligation signal (red) and the pThr73-Rab10 signal (gray) in rats treated with vehicle, PF-360 alone, rotenone alone, or rotenone + PF-360. TH, tyrosine hydroxylase, a marker of dopamine neurons (blue). (B) Quantification of pSer1292 proximity ligation signal in rats treated with vehicle, PF-360 alone, rotenone alone, or rotenone + PF-360. Symbols represent individual rats. Comparison by ANOVA with Bonferroni correction. (C) Quantification of pThr73-Rab10 signal in rats treated with vehicle, PF-360 alone, rotenone alone, or rotenone + PF-360. Symbols represent individual rats. Comparison by ANOVA with Bonferroni correction. (D) Shown is pSer129-α-synuclein immunoreactivity in rats treated with vehicle, PF-360 alone, rotenone alone, or rotenone + PF-360. (E) Quantification of pSer129-α-synuclein signal in rats treated with vehicle, PF-360 alone, rotenone alone, or rotenone + PF-360. Symbols represent individual rats from a single experiment. Comparison by ANOVA with Bonferroni correction.We reported previously that chronic rotenone treatment (10 to 14 days) leads to elevated pSer129-α-synuclein (15). Here, we found that the rats treated for only 5 days also accumulated pSer129-α-synuclein, and cotreatment with PF-360 prevented this accumulation (Fig. 5, D and E). The mechanism by which pSer129-α-synuclein accrues in response to rotenone is uncertain, but it has been suggested that phosphorylation of α-synuclein at Ser129 targets the protein for degradation by autophagy (25, 26). Both chaperone-mediated autophagy (CMA) and macroautophagy play roles in α-synuclein degradation (27, 28). Therefore, we assessed a marker for CMA, Lamp2A, which is located on lysosomes, and another marker, Lamp1, which may label late endosomes, autolysosomes, or lysosomes. There were abundant Lamp2A and Lamp1 punctae in nigrostriatal dopamine neurons from the brains of vehicle-treated rats, which were markedly lost after rotenone treatment and preserved by cotreatment with PF-360 (Fig. 6, A to E). Together, these results suggest that there may be early impairment of CMA and lysosomal function, which is downstream of LRRK2 kinase activity. To complement these pharmacological studies, we examined the effects of rotenone on pSer129-α-synuclein in wild-type and LRRK2−/− HEK-293 cells. We found that rotenone treatment caused accumulation of pSer129-α-synuclein in wild-type cells; however, there was no such accumulation in the LRRK2 null cells (fig. S4), suggesting that buildup of pSer129-α-synuclein may be LRRK2-dependent. Moreover, the rotenone-induced increase in pSer129-α-synuclein in wild-type cells was effectively blocked by PF-360 to the same extent as in LRRK2−/− cells, confirming the specificity of the PF-360 effect.
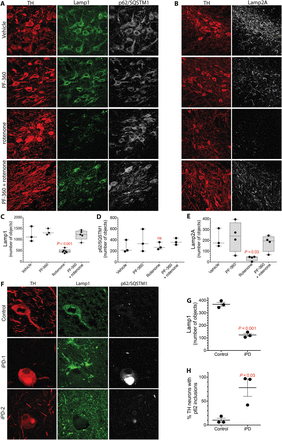
Fig. 6 Rotenone induces lysosomal and CMA defects in rat nigrostriatal dopamine neurons that are prevented by cotreatment with a LRRK2 kinase inhibitor.(A) Shown is Lamp1 and p62/SQSTM1 immunoreactivity in the nigrostriatal dopamine neurons of rats treated with vehicle, PF-360 alone, rotenone alone, or rotenone + PF-360. TH, tyrosine hydroxylase, a marker of dopamine neurons (red). (B) Shown is Lamp2A immunoreactivity in the nigrostriatal dopamine neurons of rats treated with vehicle, PF-360 alone, rotenone alone, or rotenone + PF-360. (C) Quantification of Lamp1 signal in rats treated with vehicle, PF-360 alone, rotenone alone, or rotenone + PF-360. Symbols represent individual rats from one experiment. Comparison by ANOVA with Bonferroni correction. (D) Quantification of p62/SQSTM1 signal in rats treated with vehicle, PF-360 alone, rotenone alone, or rotenone + PF-360. Symbols represent individual rats from one experiment. Comparison by ANOVA with Bonferroni correction. (E) Quantification of Lamp2A signal in rats treated with vehicle, PF-360 alone, rotenone alone, or rotenone + PF-360. Symbols represent individual rats from one experiment. Comparison by ANOVA with Bonferroni correction. (F) Lamp1 and p62/SQSTM1 immunoreactivity in the substantia nigra of a postmortem human healthy, age-matched control brain and two postmortem brains from iPD patients (iPD-1 and iPD-2). In the control brain, nigrostriatal dopamine neurons contained many small punctae of Lamp1 immunoreactivity and little detectable p62/SQSTM1. In the two postmortem iPD brains, there was loss of Lamp1 puncta and accumulation of p62/SQSTM1 into large inclusions (Lewy bodies) in nigrostriatal dopamine neurons. (G) Quantification of Lamp1 in postmortem brain nigrostriatal dopamine neurons from three healthy age-matched control subjects and three patients with iPD. Symbols represent individual brains. Comparison by unpaired two-tailed t test. (H) Quantification of p62 in postmortem brain nigrostriatal dopamine neurons from three healthy age-matched control subjects and three patients with iPD. Symbols represent individual brains. Comparison by unpaired two-tailed t test.Similar to rotenone-treated rats, there was a marked loss of Lamp1 puncta in human postmortem brain tissue from individuals with iPD (P < 0.001, unpaired two-tailed t test), and this was accompanied by an accumulation of the autophagy cargo receptor p62/SQSTM1 in Lewy bodies (P < 0.03), indicating autophagic and lysosomal dysfunction (Fig. 6, F and G). As reported by many other groups, and as seen in rotenone-treated rats, there was accumulation of pSer129-α-synuclein in the substantia nigra of postmortem brain tissue from iPD patients.In vitro experiments have shown that pSer129-α-synuclein binds to TOM20 and inhibits mitochondrial protein import; however, this has not been examined directly in human brain or in the rotenone-treated rat model of PD. Examination of human iPD postmortem brain tissue (Fig. 7, A and B) revealed a marked increase in the pSer129-α-synuclein–TOM20 proximity ligation signal (P < 0.0001, unpaired, two-tailed t test), indicating that accumulation of this specific form of α-synuclein may have toxic consequences in terms of mitochondrial protein import. Similarly, in the rotenone-treated rats (Fig. 7, C to E), the increased pSer129-α-synuclein we found at 5 days was associated with its binding to TOM20, measured as a strong pSer129-α-synuclein–TOM20 proximity ligation signal (P < 0.0001, two-way ANOVA with Bonferroni correction), as well as reduced levels and redistribution of the imported complex I subunit, Ndufs3, from mitochondria to cytosol (P < 0.0001). Cotreatment with PF-360 prevented the elevation in pSer129-α-synuclein (Fig. 5E) and, as a result, there was little binding to TOM20 (P < 0.0001 versus rotenone alone), and there was preservation of normal levels and mitochondrial localization of Ndufs3 (Fig. 7, C and E). Thus, both the accumulation of pSer129-α-synuclein and its toxic consequences appear to be downstream of LRRK2 kinase activity.Fig. 7 pSer129-α-synuclein binding to TOM20 in postmortem iPD brain tissue and in rotenone-treated rats is prevented by cotreatment with a LRRK2 kinase inhibitor.(A) Shown is the pSer129-α-synuclein (pSer129syn)–TOM20 proximity ligation signal in the substantia nigra of postmortem brain tissue from a healthy, age-matched control individual and a patient with iPD. (B) Quantification of the pSer129syn-TOM20 proximity ligation signal in eight postmortem control brains and seven postmortem iPD brains. Comparison by unpaired two-tailed t test. Symbols represent individual brains. (C) Shown is pSer129syn-TOM20 proximity ligation signal (red) and Ndufs3 immunoreactivity (gray) in the substantia nigra of the brains of rats treated with vehicle, PF-360 alone, rotenone alone, or rotenone + PF-360. In rotenone-treated rats, there was increased pSer129syn-TOM20 proximity ligation signal and a reduced amount and diffuse redistribution of the nuclear encoded and imported complex I subunit Ndufs3. These abnormalities were prevented by treatment with the LRRK2 kinase inhibitor PF-360. (D) Quantification of the pSer129syn-TOM20 proximity ligation signal in the substantia nigra of the brains of rats treated with vehicle, PF-360 alone, rotenone alone, or rotenone + PF-360. Symbols represent individual rats from a single experiment. Comparison by ANOVA with Bonferroni correction. (E) Graphical representation of the distribution and fluorescence intensity of Ndufs3 in nigrostriatal dopamine neurons in the brains of rats treated with vehicle, PF-360 alone, rotenone alone, or rotenone + PF-360. Note the loss of punctate, high-intensity staining in the rotenone-treated animals that was preserved by cotreatment with PF-360.